本文献已发布在上一期推文中,(灰色和斜体字),用于辅助理解原文
接收: 2019-12-11,Nature
作者:PCAWG Transcriptome Core Group
链接:doi.org/10.1038/s41586-020-1970-0
(仔细理解这句话是癌症中联合转录组和基因组数据分析的理论基础。即转录(组)变化的分子机制来自基因组)
(这些大多是转录组测序的研究内容,但我们往往只关注基因表达定量、差异基因鉴定、GO/KEGG等功能注释)
(这里的异质性主要指:1. 每个人的遗传差异和2. 不同取样部位、取样时间和不同细胞亚型在肿瘤组织中的差异。这些差异或异质会极大地影响肿瘤研究中的样本设置和数据分析策略。
① 肿瘤WES研究除了需要患者自己的肿瘤组织外,还需要自己的非肿瘤组织来匹配,后者是过滤掉患者独特的胚胎系统变异(即我们生来就与其他人不同DNA序列变异),否则将无法判断来自体细胞的突变 (后天获得),还是天生就有 (遗传或新突变),即:Somatic Mutation vs. Germline Mutation。公共数据库不记录患者的先天遗传变异,因此患者自身的正常组织总是需要配对。
② 在大多数情况下,体细胞突变是随机的。因此,同一肿瘤组织一般可分为不同的细胞亚群,包括不同的驱动突变,然后有不同的转录表达 (这是空间异质性。肿瘤的演变/进化研究可能涉及时间异质性,如晚期癌症转移组织或服用靶向药物一年后的耐药性突变组织等。
但总的来说,我们认为不同的器官、组织和细胞携带相同的遗传物质,即:Germline突变不受材料的影响,可以是癌症、全血、白细胞甚至口腔脱落细胞;肿瘤细胞样本的材料要复杂得多,需要考虑:正常的组织匹配、时间和空间异质性。
由于异质性的存在,需要多样化的取样方法和较大的总样本量 (即较大的患者队列),获得更可信的分子机制预测结果)
(进一步)
(我们发现了)
(还)
(总之)
(之前)
(每类肿瘤154~6个样本,见下图;平均值:44)也包含了少数转移组织(Extended Data Fig. 1, Methods, Supplementary Results, Supplementary Table 23)
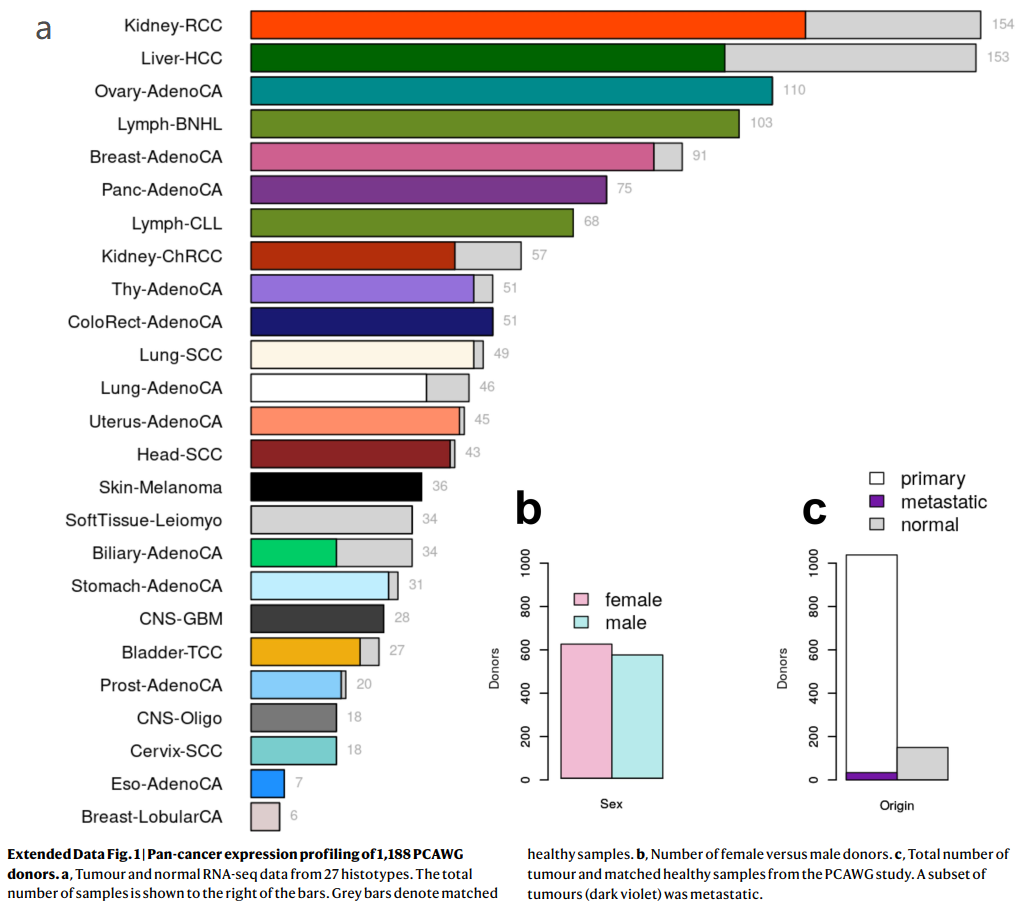
Extended Data Fig. 1 | 1,188例PCAWG捐献者的泛癌表达谱
a,来自27种组织类型的肿瘤和正常RNA-seq数据。样本总数显示在柱状图的右边。条表示匹配的健康样本。
b,女性和男性捐献者的数量。
c,来自PCAWG研究的肿瘤总数和匹配的健康样本。一组肿瘤()已转移。
S1_covars.xlsx / All_samples_cohort
Supplementary_Tables (下载):https://pan.baidu.com/s/10fTsnVYlk30T9pKIq05cHg
提取码:ysx4
Cancer-specific germline cis-eQTLs
(Extended Data Fig. 2)
(表达数量性状位点 (Expression quantitative trait locus, eQTL)是关联转录组和基因组/外显组两个组学的常用、经典方法,属于多组学研究范畴。
eQTL是一类能够影响基因表达量的遗传位点(大部分都是单核苷酸多态性,SNP),具有一定的生物学意义。迄今为止最全的eQTL数据库是GTEx。分析SNP和基因表达水平的关联度,以及SNP与基因的距离,寻找SNP调控的基因。
Expression quantitative trait loci (eQTLs) are genetic variants that influence expression levels of mRNA transcripts. Cis-eQTLs commonly refer to genetic variations that act on local genes , and trans-eQTLs are those that act on distant genes and genes residing on different chromosomes.
a) cis-eQTL, b) trans-eQTL, c) mediated/介导 trans-eQTL with a single cis-mediator, and d) mediated/介导 trans-eQTL with multiple cis-mediators. 清华大学统计科学中心,https://doi.org/10.1186/s12859-019-2651-6,BMC Bioinformatics
Identification of eQTLs can help advance our understanding of genetics and regulatory mechanisms of gene expression in various organisms. Consistent findings suggest that many genes are regulated by nearby SNPs, and the identified cis-eQTLs are typically close to transcription start sites (TSSs). In contrast to cis-eQTLs, trans-eQTL identification is much more challenging because a greater number of SNP-gene pairs are tested for trans-association. In order to achieve the same power, analysis of trans-eQTLs requires a much larger sample size and/or effect than that in the cis-eQTL analysis. However, trans-eQTLs tend to have weaker effects than cis-eQTLs.
Mediation diagram of the trans-association between rs2239804 and RPL34
Several methods have been developed to improve trans-eQTL detection, such as reducing the multiple-testing burden based on pairwise partial correlations from the gene expression data to increase power, and constructing or selecting variables to control for unmeasured confounders that may lead to spurious association
eQTL分析至少需要三个文件:第1个是样本信息文件,该文件包含样本的年龄,性别和人种等等;第2个是基因表达量文件,它表示的是每个基因在每个样本中的表达含量;第3个是基因型数据,也即每个样本的基因型数据)
Extended Data Fig. 2 | 概述:在分析中考虑的遗传变异的不同来源
a, 为了分析,使用标准eQTL方法,分别检测单等位、单核苷酸 (Mono-allelic single-nucleotide)变异 ()与 (Total gene expression)的关联。(蓝色圆点SNV,在样本中存在完全相同的基因组位置;上图的示例有)
由于SNV在队列中复发率较低SNV,在样本中完全相同的基因组位置;上图的示例有,根据它们 (例如启动子、5 ' UTR或内含子),体细胞SNV被 (Aggregated in burden categories) (例如上图的“Local somatic SNV burden/局部体细胞SNV负荷”)。
然后,获取与所有基因的,以及在每个基因水平上的总表达。通过检测与突变及表观遗传特征相关的总基因表达,来估计。
所有的窗口大小为;的窗口大小为。
b,概述:不同的数据集,及其对a中所述分析的贡献。箭头表示所执行的单个分析之间的依赖关系。
① 胚系基因型来源于匹配的 (Matched)健康全基因组测序 (WGS)样本。
② 等位基因特异性SCNAs (体细胞拷贝数改变)、突变特征和局部SNV负荷,来自于:与未受影响的 (Unaffected) WGS样本相比的肿瘤WGS (即N-T配对)。
③ ASE和总表达 (Total expression/FPKM)来自肿瘤和正常RNA-seq数据。
(Extended Data Fig. 3, Supplementary Table 1)
Supplementary_Tables / S2_eGenes_v2.xlsx / pan-analysis
Extended Data Fig. 3 | 胚系eQTL中的变异 ( variants)
(每一行的3个子图是一类肿瘤,3个图是eQTL分析常见的输出图形,主要涉及:P值、先导SNP的个数及其与TSS的距离)
(Marginal significance. P≤0.01, Bonferroni-adjusted)
(~8.4%) (Extended Data Fig. 4, Supplementary Table 3)
Fig. 1 | 与基因表达关联的胚系及体细胞SNV
a,表观遗传学路线图 (Epigenetics Roadmap)富集分析,显示泛分析/Pan-analysis的PCAWG特异性eQTLs,以及在GTEx组织中复现/Replicate的eQTLs中,跨细胞系Roadmap因素/Factos的平均倍数变化。
* :P < 0.05/25, PCAWG特异性eQTLs的单侧Wilcoxon秩和检验,校正了所使用的Roadmap因子的数量 (即25)。数据为均值和标准差.
(其它几个子图,将在后文讲解)
Somatic cis-eQTLs in non-coding regions
Extended Data Fig. 5 | 顺式突变体细胞负荷 (Cis-mutational somatic burden)
a,每种癌症类型的体细胞突变负荷总数 (Total number of somatic mutational load per cancer type)。SNV的中位数范围从甲状腺腺癌的1,139个到皮肤黑色素瘤的72,804个。
(此图也可以用于绘制肿瘤样本分类或分组后,各自体细胞突变负荷总数的分布图)
(横轴) Shared Aliquots (共享的整除数)
b,由越来越多的患者共享的反复出现的体细胞SNV的数量。一小部分 (≥86个SNV)在超过1%的队列 (≥12例患者)中均被检测到。
(此图可由变异水平的各样本的SNV矩阵 (热图)/VCF文件,统计得到)
(Extended Data Figs. 2, 5, 6)
Extended Data Fig. 6 | 按检测区域类型划分的体细胞突变率与负荷频率 ( and )
a,每个基因检测到的、体细胞突变负荷频率≥1%的、突变区域的个数;
b,每千碱基的突变率 (Mutation rate per kilobase)。
c,按所测间隔类型划分的 (侧翼区、外显子、内含子)负荷频率。
d,前导间隔 (Leading intervals, FDR≤5%)到其最近的 (左和右)间隔的距离分布 (bp),使其关联的P值下降了至少一个数量级 (显示了99%的分布)。
e,检测的所有的基因组区域 (负荷频率≥1%,n = 1,049,102),以及所观察到的FDR为5%的体细胞顺式eQTL下的567个基因组区域的分解 (Breakdown)。图中,Intronic:eGene内含子;Exonic:eGene外显子;Flank.:表示距离eGene起始和结束1Mb距离内的2kb侧翼区域;flank.intergenic:指基因组位置 (无基因注释)的侧翼区域;Flank.intronic:指与邻近基因内含子重叠的侧翼区域;Flank.others:表示与附近基因的一些注释部分地重叠的侧翼区域。
b,对基因表达水平进行方差成分分析 (Variance component analysis),显示不同种系和体细胞因素,对不同基因集的方差所占的平均比例 (Average proportion of variance explained by different germline and somatic factors for different sets of genes),包括所有因子的平均效应:
1)所有遗传因子 (包括种系和体细胞);2)体细胞拷贝数变异;3)侧翼区的体细胞变异;4)人群结构;5) cis-germline effects;6)体细胞内含子和外显子突变效应。
(可见:体细胞的内含子和外显子突变效应的解释度很小,而主要由拷贝数变异、非编码区和顺式胚系等变异所解释)
(Supplementary Table 5) (Extended Data Figs. 7, 8)
Extended Data Fig. 7 | 与遗传先导负荷 (Genic lead burden)相关联的7个体细胞eGenes的曼哈顿图
Extended Data Fig. 8 | 8个体细胞eGenes的散点图,显示先导权重负荷对基因表达残差的影响 (Plots show the effect of the lead weighted burden on the gene expression residuals (见原文的Methods) of these genes. a, CDK12. b, PI4KA. c, IRF4. d, AICDA. e, C11orf73. f, BCL2. g, SGK1. h, TEKT5
(Extended Data Fig. 9, Supplementary Table 6) (Supplementary Table 7)(Extended Data Fig. 9)
Extended Data Fig. 9 | 与存在体细胞突变负荷的侧翼间隔,有所重叠的表观基因组图谱标记 (Roadmap epigenome marks)
(P = 0.04, Fisher’s exact test) (Fig. 1c, Extended Data Fig. 8h)
Fig. 1c
c,曼哈顿图显示TEKT5基因关联的名义 (Nominal)P值 (用灰色标出),已考虑侧翼、内含子和外显子间隔。先导体细胞负荷与TEKT5表达的增加相关 (P = 1.61 × 10e-6),并与上游二价 (Bivalent)启动子重叠 (红点;注释于:81个Roadmap细胞系,包括8个胚胎干细胞,9个胚胎干细胞来源,5个诱导多能干细胞系)。
(Supplementary Table 8)
Fig. 1d, 1e
d,突变特征 (Mutational signatures, Sig)与基因表达之间的显著性关联结果总结。
顶:每1类突变特征/Signature (FDR ≤ 10%)中,关联基因的总数。
下:每1类突变特征/Signature相关的基因,其富集到的GO分类/Categories或Reactome通路 (FDR≤10%,显著性水平以颜色编码,-log10转换后的校正后的P值)。
e,仅考虑SCNAs、胚系eQTLs、编码和非编码突变,AEI (Allelic expression imbalance,非平衡等位基因表达)存在的标准效应 (Standardized effect)大小。数据是对效应大小的估计和标准误的估计。
-- 未翻译完,更多内容请查看原文;下文主要涉及:摘要、方法和部分图形解读 --
Fig. 2 | 体细胞突变对选择性剪接的位置特异性影响 (Position-specific effect of somatic mutations on alternative splicing)
a,顶部,外显子-内含子连接 (Exon–intron junctions)附近,及与外显子跳过事件 (Exon-skipping event)相关的分支位点 (Branch sites)的突变比例。具有相关剪接变化的突变是指其中:The percentage spliced in-derived |z-score| is ≥ 3 (图中的深蓝色)。星号:Intron positions significantly enriched for splicing changes relative to background based on a permutation test. *P < 0.05, **P < 0.01, ***P < 0.001。底部: sequence motifs of regions。
Fig. 2b, 2c
b,肿瘤抑制基因STK11的。图的上方,对于携带变异 (Alternative/替代)等位基因的供体,基因的某部分的RNA-seq的Reads覆盖显示为,而对于携带参考等位基因的随机供体 (Random donor with reference allele)则显示为。盒式外显子事件 (Cassette exon event)显示在图的下方。
c, Enrichment of SINE elements in SAVs (Splicing-associated variants,剪接相关变异) compared to sequence background (BG). Shown for SINE elements overlapping in sense (middle) and antisense (right) directions.
Fig. 3 | 与RNA融合相关的结构重排
a,所有检测到的和新的融合的数量,及其与癌症普查 (Census)基因的重叠部分。b、桥接融合示意图。桥接融合是由连接两个基因的第三个基因组片段形成的复合融合。在每种情况下,只描述了一种可能的基因组排列顺序,断点被突出显示为“闪电”。
Fig. 4 | 影响肿瘤的DNA和RNA变化的全局视图
a, 不同组织类型的不同改变的中位数. Histotypes are ordered by hierarchical clustering based on the pattern of different types of alteration. Alt., alternative; non-syn, non-synonymous. Cancer-type abbreviations are listed in Supplementary Table 23.
b, c, Circular representations of the selected genes significantly co-occurred with B2M (b) and PCBP2 (c). Connecting lines indicate the specific types of co-occurrence of alteration pairs. 内部直方图显示不同颜色的不同DNA/RNA变化类型的发生频率。
d, 所有癌症体细胞突变目录 (COSMIC)的癌症普查基因,或PCAWG驱动基因,在RNA和DNA水平的改变中、存在频繁和异质性地改变。黄条:DNA水平发生改变的样本比例,绿条:RNA水平发生改变的样本比例。(二者呈现相反的趋势,可以这么理解:肿瘤中如果一个基因已经发生了突变,则其表达与否,是次要影响因素,后者不再受癌症演变的选择;反之亦然。有些基因注定是驱动突变 (如TP53),另外一些基因则是“被动表达 (如GAS7)”,即驱动突变引起的对其它一系列基因表达调控的影响)。中间一栏:该基因观察到的每种变异类型的比例。
e, 在我们发现的显著地重复出现的基因的列表中的癌症基因的富集 (The enrichment of cancer genes within our list of significantly recurrent genes)。
接收: Nature Communications
时间/作者:2021/丹麦奥胡斯大学医院分子医学系
链接:doi.org/10.1038/s41467-021-22465-w
(临床上,高危NMIBC手术后经膀胱辅助灌注 (Bacillus Calmette–Guérin, BCG)以根除残留疾病,从而减少复发和进展的频率)
Fig. 1 .
Consensus matrix for four clusters. Samples are in both rows and columns and pairwise values range from 0 (samples never cluster together; white) to 1 (samples always cluster together; dark blue). (样本的相关性矩阵,发现聚集为4类)
Comparison between the three UROMOL2016 transcriptomic classes and the UROMOL2021 four-cluster solution (76% of tumors in UROMOL2016 class 1 remained class 1, 92% of tumors in UROMOL2016 class 2 remained class 2a/2b and 67% of tumors in UROMOL2016 class 3 remained class 3). (样本前后分类、聚集的比较)
Kaplan–Meier plot of progression-free survival (PFS) for 530 patients stratified by transcriptomic class. (以分组的转录组聚集分类,做无进展生存曲线;四条曲线分别对应4种分类)
Kaplan–Meier plot of recurrence-free survival (RFS) for 511 patients stratified by transcriptomic class. (同上,无复发生存期 生存曲线)
, Clinicopathological information and selected gene expression signatures for all patients stratified by transcriptomic class. Samples are ordered after increasing silhouette score within each class (lowest to highest class correlation). CIS carcinoma in situ, EORTC European Organisation for Research and Treatment of Cancer, EAU European Association of Urology, MIBC muscle-invasive bladder cancer, EMT epithelial-mesenchymal transition. (转录组分类的,所有患者的临床病理信息、及选定的基因表达特征,二者的信息映射。样本在每个类别中增加轮廓分数后排序(从最低到最高类别相关性)。CIS原位癌,EORTC欧洲癌症研究和治疗组织,EAU欧洲泌尿学协会,MIBC肌肉浸润性膀胱癌,EMT上皮-间质转化) (比如EMT基因集合,在各个样本中的表达值做加和?)
RNA-based immune score and immune-related gene expression signatures for all patients stratified by transcriptomic class. (转录组分类的所有患者的RNA免疫评分和免疫相关基因表达特征)
Regulon activity profiles for 23 transcription factors. Samples are ordered after increasing silhouette score within each class (lowest to highest class correlation). Regulons (rows) are hierarchically clustered. (23个转录因子的调控活性图谱。样本在每个类别中增加轮廓分数后排序(从最低到最高类别相关性)。规则(行)是层级聚类的)
Regulon activity profiles for potential regulators associated with chromatin remodeling. The most-upregulated regulons within each class are shown. Regulons are hierarchically clustered. P-values were calculated using two-sided Fisher’s exact test for categorical variables, Kruskal–Wallis rank-sum test for continuous variables and two-sided log-rank test for comparing survival curves. Source data are provided as a Source data file. (与染色质重塑相关的潜在调控因子的调控活性谱。每个类别中最受限制的规则显示出来。规则是层级聚类的。P值的计算采用分类变量的双侧Fisher精确检验,连续变量的Kruskal-Wallis秩和检验,生存曲线的比较采用双侧log-rank检验。源数据作为源数据文件提供)
图2 NMIBC中
根据基因组类别 () 1-3分层的473个肿瘤的图。增益(增益+高平衡增益)和损失(损失+高平衡损失)汇总在染色体带面板的左侧。EORTC欧洲癌症研究与治疗组织,EAU欧洲泌尿外科协会,MIBC肌肉浸润性膀胱癌。
426例按基因组分类的无进展生存期(PFS) Kaplan-Meier图。
399例按基因组分类的患者无复发生存期(RFS) Kaplan-Meier图。
EORTC高危评分(n = 163)按基因组分类分层的患者的PFS Kaplan-Meier图。p值的计算采用双侧log-rank检验。源数据作为源数据文件提供。
Fig. 3 Genomic alterations associated with transcriptomic classes.
Genomic classes (GCs) compared to transcriptomic classes (n = 303). 。
compared to GCs. Colors indicate transcriptomic classes.
Kaplan–Meier plot of progression-free survival () for 154 patients (including only class 2a and 2b tumors) stratified by .
Number of according to transcriptomic classes.
Landscape of genomic alterations . Samples are ordered after the combined contribution of the APOBEC-related mutational signatures. Panels: RNA-derived mutational load, (inferred from 441 tumors having more than 100 single nucleotide variations), selected RNA-derived mutated genes, in selected disease driver genes (derived from SNP arrays). Asterisks indicate p-values below 0.05. Daggers indicate BH-adjusted p-values below 0.05.
of to data from 38 patients for 11,016 mutations in all genes, 280 mutations in the genes most frequently mutated or differentially affected between the classes (n = 82, Supplementary Fig. 5b) and 93 mutations in 19 selected bladder cancer genes (Fig. 3e). Only mutations with > 10 reads in tumor and germline DNA were considered and a mutation was called observed when the frequency of the alternate allele was above 2%.
(including TP53, ATM, BRCA1, ERCC2, ATR, MDC1).
RNA-based to GCs.
RNA-derived to GCs.
Relative contribution of the transcriptomic class.
P-values were calculated using two-sided Fisher’s exact test for categorical variables, Kruskal–Wallis rank-sum test for continuous variables and twosided log-rank test for comparing survival curves. For all boxplots, the center line represents the median, box hinges represent first and third quartiles and whiskers represent ± 1.5× interquartile range. Source data are provided as a Source data file.
Fig. 4 . a Multiplex immunofluorescence staining with Panel 1 (CD3, CD8, and FOXP3) of tumors with high- and low immune infiltration with magnifications of T helper cells (CD3+, CD8− and FOXP3−), a cytotoxic T lymphocyte (CTL; CD3+, CD8−, FOXP3−) and a regulatory T cell (Treg; CD3+, CD8− and FOXP3+). Yellow dashed lines divide the tumor tissue into parenchymal and stromal regions. Scale bar: 20 µm. All protein measurements were performed once for each distinct sample. b Spatial organization of immune cell infiltration and antigen recognition/escape mechanisms (MHC class 1 and PD-L1) with associated data for genomic class, transcriptomic class, and recurrence rate. The immune cells and immune evasion markers are defined as the percentage of positive cells in the different regions (stroma and parenchyma) and normalized using zscores, (1) z ¼ ðxμÞ σ . Columns are sorted by the degree of immune infiltration into the tumor parenchyma in descending order from left to right. c Immune infiltration stratified by transcriptomic class. Immune infiltration is defined as the percentage of total cells in the parenchyma classified as immune cells. The p-value was calculated using two-sided Wilcoxon rank-sum test. d Immune infiltration stratified by recurrence rate. The p-value was calculated by the one-sided Jonckheere–Terpstra test for trend. e Kaplan–Meier plot of recurrence-free survival (RFS) for patients with tumors with few genomic alterations (GC1 + 2) stratified by immune infiltration. P-value was calculated using two-sided log-rank test. f Distribution of CK5/6 and GATA3 positive carcinoma cells stratified by transcriptomic class. Each column represents a patient. The p-value reflects the difference in CK5/6 expression across classes and was calculated by chi-squared test. For boxplots, the center line represents the median, box hinges represent first and third quartiles and whiskers represent ± 1.5× interquartile range. Source data are provided as a Source data file.
Fig. 5 . a Overview of hazard ratios calculated from univariate Cox regressions of progressionfree survival using clinical and molecular features. Black dots indicate hazard ratios and horizontal lines show 95% confidence intervals (CI). Asterisks indicate p-values below 0.05 and the sample sizes, n, used to derive statistics are written to the right. CIS carcinoma in situ, EORTC European Organisation for Research and Treatment of Cancer, EAU European Association of Urology. b Receiver operating characteristic (ROC) curves for predicting progression within 5 years using logistic regression models (n = 301, events = 19). Asterisks indicate significant model improvement compared to the EORTC model (Likelihood ratio test, BH-adjusted p-value below 0.05). AUC area under the curve, CI confidence interval. c Summary characteristics of the transcriptomic classes. Molecular features associated with the classes are mentioned, and suggestions for therapeutic options with potential clinical benefit are listed. MIBC muscle-invasive bladder cancer, EMT epithelial-mesenchymal transition, CTLs cytotoxic T lymphocytes. Source data are provided as a Source data file.
Fig. 6 . a Summary of classification results and stage distribution for all tumors, tumors with microarray data and tumors with RNA-Seq data (1228 tumors were classified in total and 1225 of these were assigned to a class). b Association of tumor stage, tumor grade and FGFR3 and TP53 mutation status with transcriptomic classes. P-values were calculated using two-sided Fisher’s exact test. c Kaplan–Meier plot of progression-free survival (PFS) for 511 patients stratified by transcriptomic class. The p-value was calculated using two-sided logrank test. d Association of regulon activities (active vs. repressed status) with transcriptomic classes in the UROMOL cohort (including samples with positive silhouette scores, n = 505) and transcriptomic classes in the independent cohorts (pooled). The heatmap illustrates BH-adjusted p-values from two-sided Fisher’s exact tests. e Pathway enrichment scores within transcriptomic classes in the UROMOL cohort (including samples with positive silhouette scores, n = 505) and transcriptomic classes in the independent cohorts (pooled). Asterisks indicate significant association between pathway and class (one class vs. all other classes, two-sided Wilcoxon rank-sum test, BH-adjusted p-value below 0.05). Triangles indicate direction swaps of pathway enrichment in the independent cohorts compared to the UROMOL cohort. GSVA gene set variation analysis. Source data are provided as a Source data file.
日期: 2021
期刊:Cancer Cell Int (IF=6.5)
链接:doi.org/10.1186/s12935-021-02049-w
Fig. 1 Landscape of somatic mutation profiles in HCC samples. Mutation information of each gene in each sample was shown in the waterfall plot, where different colors with specific annotations at the bottom meant the various mutation types. The barplot above the legend exhibited the number of mutation burden. Cohort summary plot displaying distribution of variants according to variant classification, type and SNV class. Bottom part (from left to right) indicates mutation load for each sample, variant classification type. A stacked barplot shows top ten mutated genes. (Rainfall plot of TCGA HCC sample TCGA−UB−A7MB−01A−11D−A33Q−10. Each point is a mutation color coded according to SNV class.) (Transition and transversion plot displaying distribution of SNVs in HCC classified into six transition and transversion events. Stacked bar plot (bottom) shows distribution of mutation spectra for every sample in the MAF file.
Fig. 2 Construction of of HCC samples.
Sample dendrogram and clinical-traits heatmap was plotted. Selection of the soft threshold made the index of scale-free topologies reach 0.90 and analysis of the average connectivity of 1–20 soft threshold power. TMB-related genes with similar expression patterns were merged into the same module using a dynamic tree-cutting algorithm, creating a hierarchical clustering tree. Heatmap of the correlations between the modules and TMB value (traits). Within every square, the number on the top refers to the coefficient between the TMB level and corresponding module, and the bottom is the P value
Fig. 3 Differential analysis of in high- and low-TMB groups and enrichment pathway annotation. Volcano plot was delineated to visualize the . Red represented upregulated and green represented downregulated. Heatmap of top 40 was drawn to reveal different distribution of expression state, where the colors of red to blue represented alterations from high expression to low expression. Venn diagram of the hub genes from WGCNA blue module and . Pathway enrichment analyses of TMB hub genes. Gene Ontology (GO) enrichment analysis of : biological processes (BP), cellular components (CC) and molecular function (MF). KEGG enrichment analysis of naïve B cells-related genes.
Fig. 4 (Validation of the prognostic risk signature in discovery group). Heatmap presents the of three hub genes in each patient. (Distribution of multi-genes signature risk score). The and interval of HCC patients. Kaplan–Meier curve analysis presenting difference of overall survival between the high-risk and low-risk groups. (Distribution of somatic mutation count). (Univariate Cox regression analyses of overall survival). (Multivariate Cox regression analyses of overall survival).
Fig. 5 预后风险特征的临床意义 (Clinical significance of the prognostic risk signature). (Heatmap presents the distribution of clinical feature and corresponding risk score in each sample. Rate of clinical variables subtypes in high or low risk score groups). Age, Gender, D WHO grade, E clinical stage, F T status, G N status and H M status
接收: Nature (Letter)
时间:2018
链接:doi:10.1038/nature25795
Extended Data Figure 1 | Cohort description and workflow. a, Venn diagram of samples analysed by whole-exome (WES), whole genome (CGI) and whole transcriptome (RNA-seq) sequencing in this cohort.